Poster 6 - Sofija Vuckovic, Vascular Surgery
MMK Department's Day 2024
Sofija Vuckovic, Vascular Surgery
Title: Integration of genetic and clinical data with single-cell transcriptomics of atherosclerotic plaques improves patient risk stratification and identifies ARNTL as a regulator of smooth muscle cell proliferation
Background
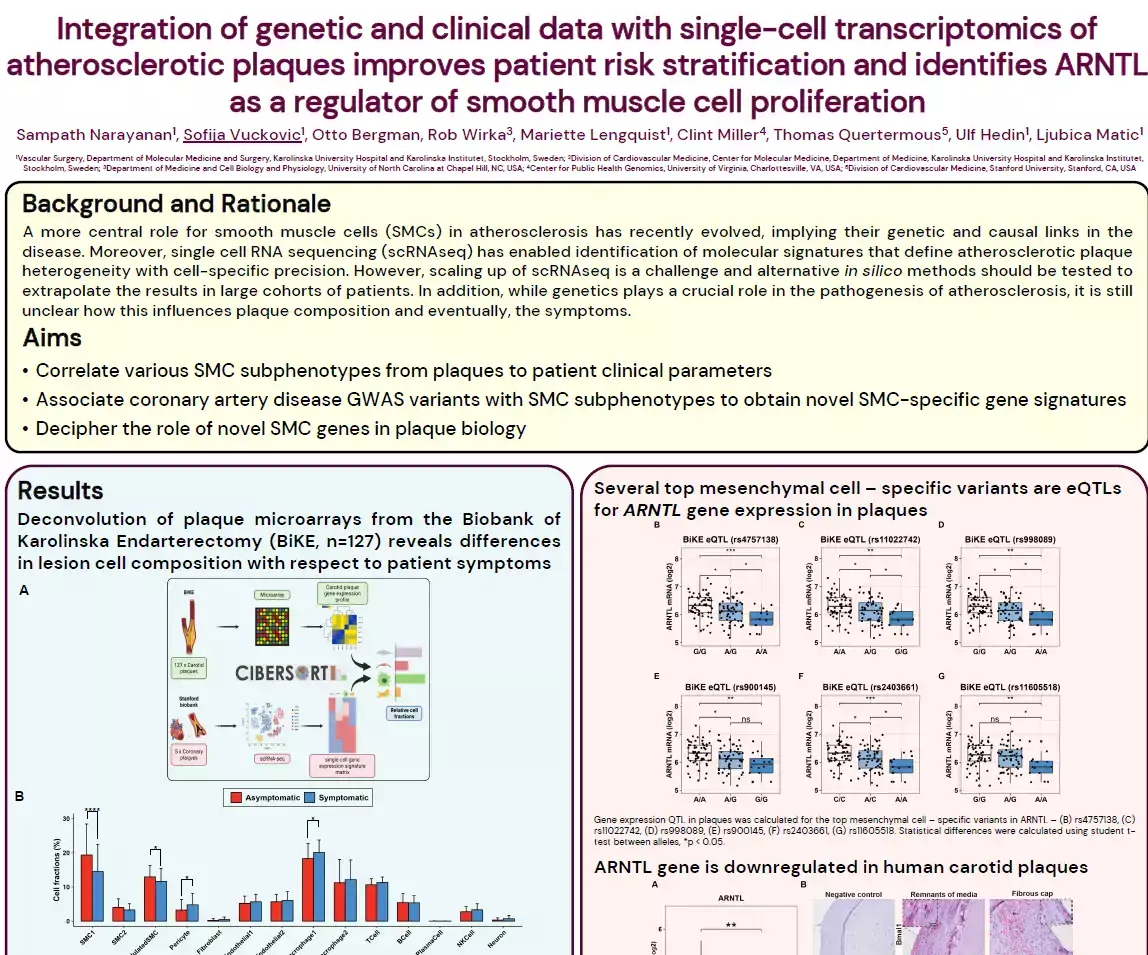
Single cell RNA sequencing (scRNAseq) of atherosclerotic lesions has been used to identify multiple cell populations of mesenchymal origin in plaques, including various smooth muscle cell (SMC) subphenotypes, but it is unknown how their ratios relate to patient clinical parameters and genetics. We correlated mesenchymal cell populations in atherosclerotic plaques with major coronary artery disease (CAD) genetic variants and performed functional analyses to identify SMC markers involved in atherosclerosis.
Methods and Results
Bioinformatic deconvolution was performed on bulk microarrays from carotid plaques in the Biobank of Karolinska Endarterectomies (BiKE, n=127) using public plaque scRNAseq data, and associated with patient clinical and follow-up information. Lesions from symptomatic patients had higher fractions of type 1 macrophages and pericytes, but lower fractions of classical and modulated SMCs compared to asymptomatic ones, particularly in females. Clustering based on plaque cell fractions revealed three distinct patient groups, with relative differences in their stability profiles and associations to stroke, even during long-term follow-up. Quantitative trait loci (QTLs) analyses were performed to predict the effect of CAD-associated genetic variants on mesenchymal cell fractions (cfQTLs) and gene expression (eQTLs) in plaques (BiKE). This revealed SNPs associated with plaque mesenchymal cell fractions upstream of the circadian rhythm gene ARNTL. In vitro silencing of ARNTL in human carotid SMCs decreased proliferation.
Conclusions
This study shows the potential of combining scRNAseq data with orthogonal clinical, genetic, and transcriptomic data for improved risk stratification. Our analyses revealed that variants in ARNTL may influence SMC ratios and function.